

Ema padece una atrofia muscular espinal y su familia requiere la solidaridad de todos para recaudar 2 millones de dólares para comprar los medicamentos.
El 28 de abril de 2020 fue el día más feliz para Natali Torterola (31) y Enzo Gamarra (30). Tras varios meses de espera, Emma -su primera hija- llegó al mundo. La pequeña nació en Resistencia, Chaco, dos días antes de la cesárea y se fueron a casa al otro día. Pero en septiembre, uno de los estudios que le realizaron a la bebé empañó tanta alegría.
A Emma le detectaron una enfermedad genética que le impide moverse y respirar con facilidad. Su vida, por estos días, se traduce en una lucha de a tres y a contrarreloj. La única esperanza es un medicamento que no se encuentra en el país y por el cual deberán recaudar, antes de que cumpla los dos años, USD 2.125.000.
«En principio fuimos a la pediatra para saber si era normal los pocos movimientos de ella y nos dijo que podría ser vaga, después fuimos a una kinesióloga pensando que era falta de estímulo y de ahí nos derivaron a una neuróloga infantil donde se le tomó una muestra que se envió a Estados Unidos. A los 21 días llegaron los resultados y nos informaron que Emmita tenía AME 1», contó el padre de la pequeña a Minuto Uno
La AME es una afección de origen genético que se caracteriza por la debilidad y la reducción muscular que se origina por la falta de movimientos. Un niño con atrofia muscular espinal recibe una copia alterada del gen SMN1 de cada uno de sus padres.
“Cuando nos enteramos de la enfermedad se nos vino el mundo abajo, pero decidimos que Emmita lleve la enfermedad lo mejor posible. Empezamos kinesiología, pileta para que ella haga los ejercicios y en ningún momento paramos. Ahora estamos en esta nueva etapa y estamos peleando, y esperamos que le va hacer muy bien a Emma y a todos los chicos AME», aseguró Gamarra.
«Lo que hace esta enfermedad es que no pueda sentarse, sostener su cabecita y levantar sus manitos. Y lo que más le afecta es la parte respiratoria», sostuvo Gamarra.
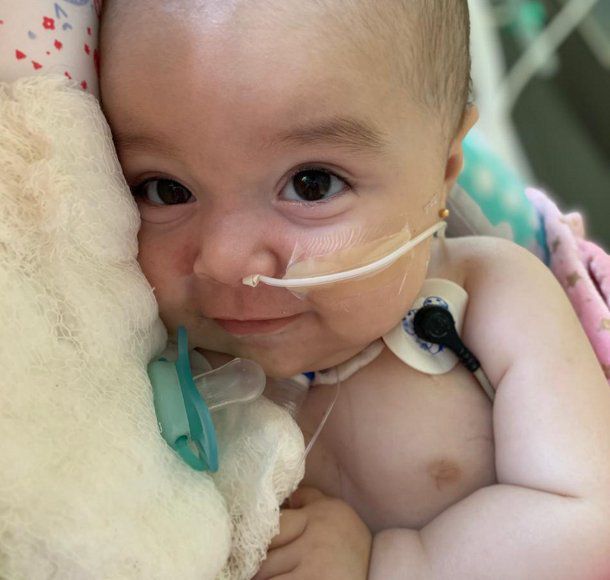
Hace un mes Emma sufrió un paro cardiorrespiratorio y fue derivada del pediátrico de Resistencia al Sanatorio Güemes, ubicado en el barrio porteño de Almagro. «Acá la tuvieron que hacer una traqueotomía porque no soportó el vuelo. Por eso decidimos movernos y poner en marcha la campaña. Esta enfermedad no le permite moverse. Tampoco respirar bien. Y su cuadro puede agravarse porque los niños con AME 1 no viven más de dos años”, contó el padre de la pequeña.
«Actualmente está con un tratamiento que se llama Spinraza, es muy nuevo, y lo que hace es alentece la enfermedad», agregó.
Hace poco conocieron un reciente tratamiento que aún no llegó al país. «El Zongensma, es otra modalidad y nos da otra esperanza para Emmita, porque actúa sobre el gen que le falta», precisó. Pero debe comenzar lo más pronto posible porque su cuadro puede agravarse, ya que los niños con AME 1 no viven más de dos años.
El medicamento no está aprobada por Anmat (Administración Nacional de Medicamentos, Alimentos y Tecnología Médica), por eso debe ser adquirido en Estados Unidos y tiene un valor de 2.125.000 dólares. El fármaco, aprobado por la FDA (Administración de Medicamentos y Alimentos) en mayo de 2019 y por la EMA (Agencia Europea de Medicamentos) en junio de este año, es desarrollado por la compañía AveXis.

Según explicó Gamarra, el medicamento se introduce por vena durante una hora y media y se da una vez única vez en la vida. «El tratamiento se aplica intratecal, primero se le ponen 4 dosis y después son dosis cada 4 meses. Para que la obra social apruebe esas dosis a Emma se le hace una evaluación y si progresa se le sigue dando la dosis, sino progresa no se le da más».
“En lugar de trabajar en el gen SMN2, como Spinraza, Zolgensma reemplaza el gen SMN1 faltante o defectuoso. Aunque Spinraza repara el gen SMN2, el gen SMN2 todavía produce menos proteína funcional que el gen SMN1. Dado que Zolgensma reemplaza el gen SMN1 faltante o defectuoso, esto proporciona a los pacientes una proteína SMN completamente funcional. Al ver realidades de niños con ambos tratamientos nos señalan que Zolgensma les cambió la vida, lograron muchos más hitos y esperanza de vida”, explicaron los padres de la pequeña en capaña.
Emma se encuentra en cama, con traqueotomía, y sus padres esperan el alta para llevarla a casa y seguir con el tratamiento. «Ella está despierta no la duermen, se sonríe todo el día, así que con esa sonrisita creemos que estamos haciendo bien las cosas y vamos a seguir para delante», manifestó Gamarra.

Junto a familiares lanzaron la campaña solidaria Todos con Emmita para recaudar los fondos necesarios para acceder al medicamento antes de que cumpla los dos años.
Además, abrieron un página (www.todosconemmita.com) en la que cuentan más de la enfermedad. «Allí contamos cómo nos cuenta y tal vez pueda ayudar a otros padres, ya que cuanto más temprano se descubre más esperanza hay», concluyó el padre de Emma.


